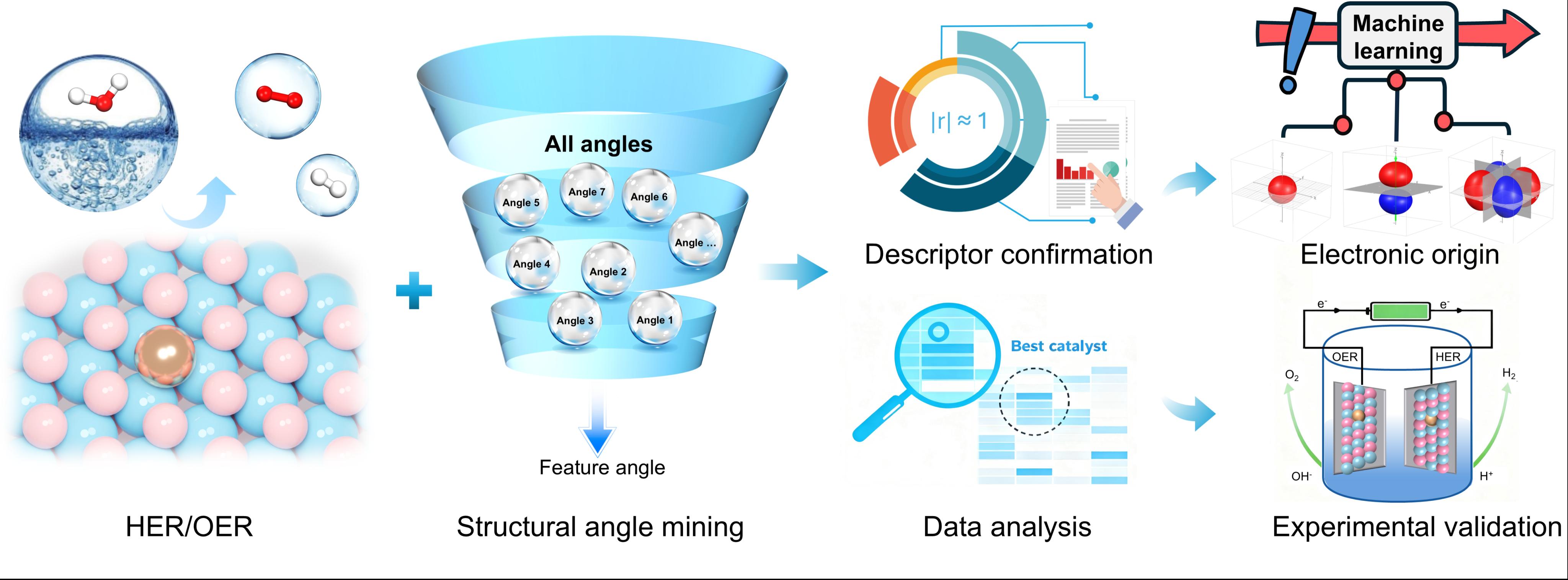
单原子催化剂(SACs)因其极高的原子利用率和可调控的电子结构,成为电催化制氢领域的前沿热点。然而,如何建立从原子级几何结构到催化性能之间的定量构效关系,一直是该领域的核心难题。传统的电子结构描述符(如d带中心)虽具预测能力,却难以转化为实验可操作的合成指导;而常规的结构描述符(如键长、配位数)又难以捕捉多维度的结构畸变信息。
近日,我院林森教授课题组与中国科学院大连化学物理研究所林坚研究员团队合作,在 《Chemical Science》 期刊上发表研究论文,首次提出了数据驱动的“角度描述符”策略,系统揭示了单原子掺杂二维过渡金属硫属化合物(TM1@MX2)中隐藏的几何设计规则。

研究团队通过对数十万种掺杂构型的高通量密度泛函理论(DFT)计算,在原子层面阐明了“外围几何调控”这一核心机制。令人意外的是,催化活性并非由掺杂原子局域配位环境主导,而是与外围配位壳层的长程角畸变密切相关。其中,第二壳层硫原子角度∠S1S23S8(φHER)对析氢反应(HER)活性的预测相关性(|r| = 0.85)显著优于掺杂原子中心角。该角度描述符框架还成功迁移至析氧反应(OER),最优角度描述符∠S8Mo52S36(φOER)与过电位呈现强相关性(|r| = 0.91)。更重要的是,该框架在四种不同MX2基质(MoS2、WS2、MoSe2、WSe2)及金属空位体系中均表现出优异的预测能力,展现了良好的普适性。针对传统d带中心理论在SACs中的失效问题,团队进一步通过SISSO算法构建了多维复合电子描述符,成功建立了结构畸变、电子结构与催化活性三者之间的内在关联。
该工作不仅首次建立了单原子催化剂在HER/OER反应中的原子级“角度-活性”构效关系,更重要的是,它成功展示了数据驱动的几何描述符如何精准预测并指导实验合成,实现了从原子尺度结构设计到宏观催化性能的跨越。研究成果提出的“外围几何调控”范式,为理性设计高性能、低成本的单原子电催化剂提供了全新的理论工具。
相关工作近期发表在 Chemical Science 上(doi.org/10.1039/D6SC01740A)。该研究得到了国家自然科学基金国际(地区)合作项目的资助。




 书记信箱
书记信箱 院长信箱
院长信箱